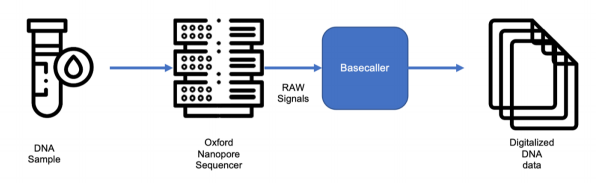
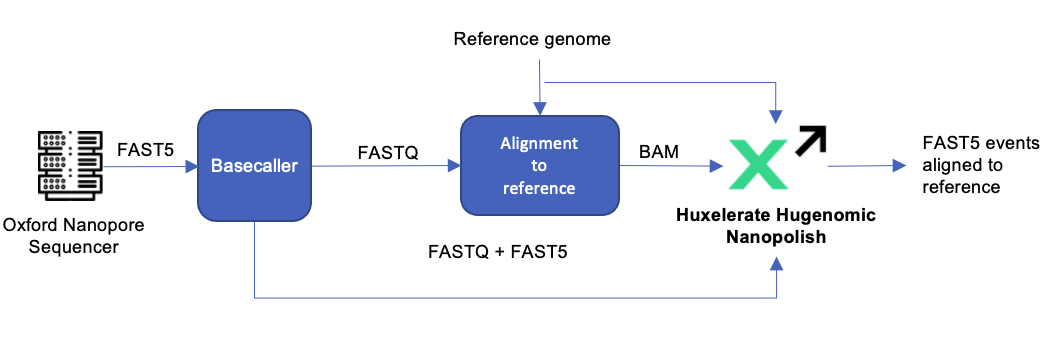
Nanopolish is a software package for signal-level analysis of Oxford Nanopore data that exploits the raw signals provided in output by the sequencing machine, to calculate an improved consensus sequence for a draft genome assembly, detect base modifications, call SNPs and indels with respect to a reference genome and more.
In this document, we will focus on a specific module called eventalign, that allows to compare the raw signals provided as output of the sequence machine, to the basecalled data, aligning signal data emitted by a nanopore machine to a reference genome, in contrast to most approaches which align two DNA sequences to each other.
The eventalign tool exploits a Hidden markov Model (HMM) to align signal data from the nanopore sequencing machine to a reference to improve the accuracy of the sequence obtained by the basecaller.
The probabilistic model is used to accomplish the task of calculating the probability of observing an event sequence (a portion of the signal provided in output from the sequencer) given a known DNA sequence (the basecalled sequence).
The HMM used within nanopolish is a profile Hidden Markov Model and helps in calculating the probability of a sequence of events, given a known sequence. Nanopolish handles huge amount of long reads and signals coming from the sequencer.